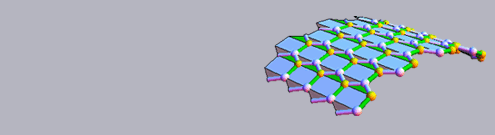
ABSTRACT One of the main goals pursued in our Laboratory in the last years was the determination of the structural features and conformational equilibria of oligopeptides and bioactive pseudopeptides in solution, making use of a technique less powerful than NMR but with a high potentiality. This method combines the results of FRET (fluorescence resonance energy transfer) measurements with the theoretical approach of molecular mechanics calculations. Where long-range energy transfer occurs, one can determine both the efficiency of the process (that depends on both the interprobe distance and mutual orientation) and population of the species in solution exhibiting that efficiency. Once the structure of the main chain has been evaluated by IR and CD spectral results, the deepest energy minimum conformers can be obtained by molecular mechanics, so that the theoretical interprobe distance and orientation in the sterically most favored structures and their relative population can be compared with those experimentally obtained by fluorescence measurements. In this way, the old problem of the orientation parameter, usually taken as <κ2> = 2/3 (average isotropic value) even when folded peptides or proteins are investigated, can be theoretically resolved. Where comparison between calculated and measured quantities is successful, a good piece of structural information is obtained, otherwise the experimental results are used as constraints for the structural calculations. By employing this technique, we were able to determine, with a high degree of confidence, more than fifty structures of different peptides or bioactive pseudopeptides in solution (mainly methanol or water/methanol). Some of these structures were later confirmed by X-ray diffraction data in the solid state. Examples of the application of this tecnnique are reported and discussed.
Buy this Article
|